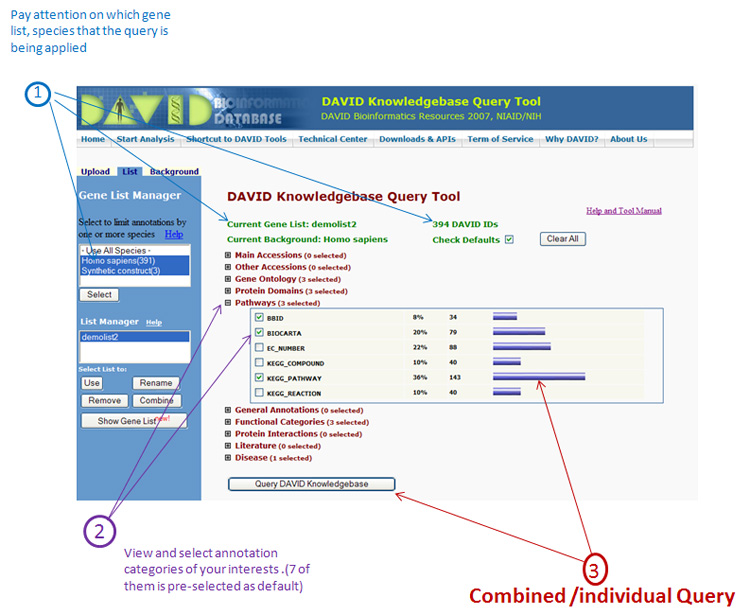
DAVID Knowledgebase
Due to the complex and distributed nature of biological research, our current knowledge is spread over many redundant databases maintained by independent groups. One gene could have different identifiers within one, or many, databases. Similarly, the biological terms associated with different gene identifiers for the same gene could be collected in different levels across different databases. Most gene functional annotation databases are in a gene-associated format, i.e. annotation contents usually associate with corresponding gene or protein identifiers . Such a format provides an opportunity to integrate heterogeneous annotation resources through their common gene identifiers. However, there are dozens of types of gene or protein sequence identifiers that are redundant across several independent groups, such as GenBank Accession; GenBank ID; RefSeq Accession; PIR ID; PIR Accession; UniProt ID; UniProt Accession; Affymetrix Probe ID; etc. The major challenge of integration comes from the weak cross-reference of different types of gene identifiers used by different functional annotation databases.
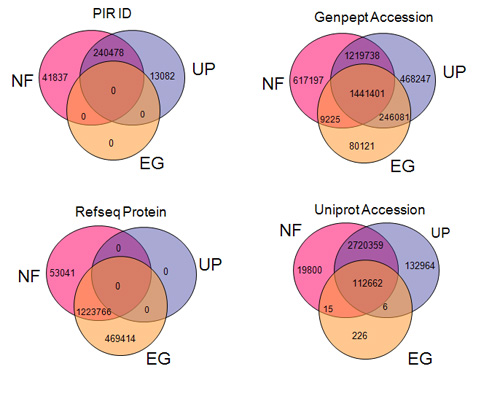
Figure:
The poor coverage and overlap of different types of protein identifiers
across
independent resources. As examples, four popular types of protein
identifiers
(PIR ID, UniProt Accession, RefSeq Protein, and GenPept Accession) are
only
covered partially by NCBI Entrez Gene (EG), UniProt UniRef100 (UP), and
PIR
NRef100 (NF). The DAVID gene collects and integrates all of them for
better
coverage and integration.
DAVID Gene Concept: DAVID
gene is a secondary gene cluster used to hold all different types
of gene IDs belonging to the same gene. Each unique gene has a
unique DAVID gene ID. DAVID Gene is conceptially
equivalent
to Entrez Gene, but with much broader data coverage cross most, if not
all, of well known bioinformatics systems.
An Example: A DAVID gene constructed by a single-linkage algorithm
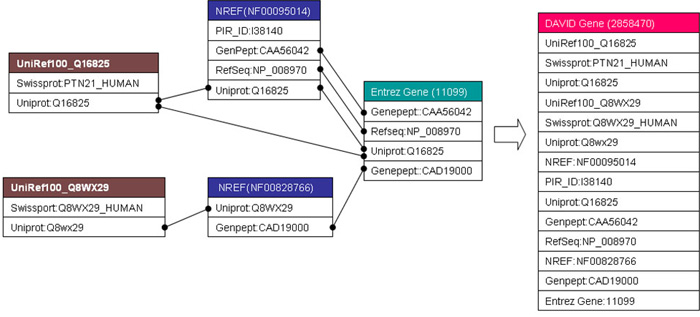
Figure: Two UniRef100 clusters, two NRef 100 clusters, and one Entrez Gene cluster were systematically found sharing one or more protein identifiers with each other. The single linkage rule can further iteratively agglomerate them as a whole into one DAVID Genegene. Thus, for this particular example of tyrosine-protein phosphatase non-receptor type 21 (PTPN21), the resulting DAVID Gene is able to integrate all gene/protein identifiers more comprehensively as compared to each original gene cluster.
Results: The process collects ~50 million individual gene/protein identifiers representing 22 identifier types, which are eventually agglomerated into over 3.7 million DAVID genes, for over 90,000 species.
DAVID Knowledgebase: After the annotations are assigned to DAVID Genes, the annotations plus DAVID Genes are called DAVID Knowledgebase.
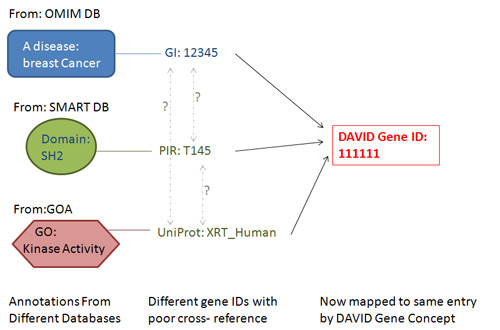
Figure: Under DAVID Gene Concept, most major types of gene identifiers can be translated to a corresponding DAVID gene identifier. Thus, as long as annotation data are in gene-associated format, the heterogeneous annotation contents have a much better chance of being integrated by the common DAVID gene identifier, thus improving the integration of annotation contents as a whole.
Results: The DAVID Knowledgebase collects a wide range of annotation contents from dozens of databases including: Gene Ontology; Protein Domains; Bio-pathways; Gene Expression; Disease Association; PubMed; Protein-Protein interactions; Affymetrix; Gene General Features; NCI Thesaurus; Panther Family; and more.
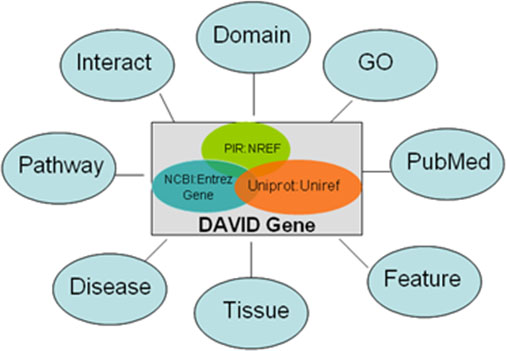
Figure: Illustration of the heterogeneous functional annotation sources integrated by DAVID genes. As long as they are in a gene-associated format, any functional annotation data sources can be linked by the common DAVID genes. Thus, a large collection of heterogeneous annotation sources can be integrated and fully cross-referenced.
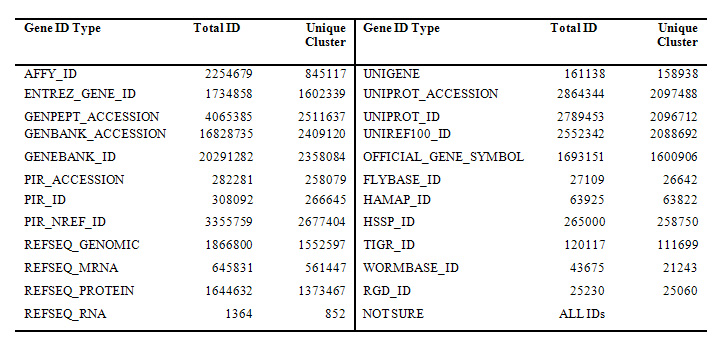
More than 20 types of gene identifers were comprehensively collected by DAVID Knowledgebase

The wide-range collection of heterogeneous functional annotations in the DAVID Knowledgebase. Over 40 functional categories from dozens of independent public sources (databases) are collected and integrated into the DAVID Knowledgebase
DAVID Knowledgebase is Organized into Pairwise
Text files.
An Example: to query data from pairwise text
formated
files in DAVID Knowledgebase
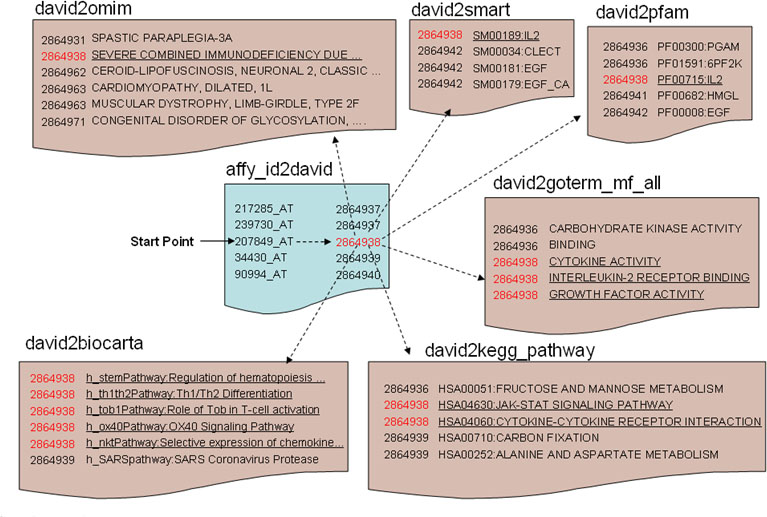
The DAVID Knowledgebase in a simple pairwise text format centralized by DAVID gene identifiers. Each independent annotation source and gene identifier system is separated into independent files in the same pairwise format of “did-to-annotation.” For this example, a user starts with Affymetrix identifier(affy_id) 207849_at (IL2). The first step is to obtain the corresponding DAVID gene identifier (2864938). Then, with this DID (red), the annotation terms of interest (underlined) in different source files (OMIM, SMART, Pfam, GO Molecular Function, KEGG Pathway, BioCart Pathway, etc.) can be queried sequentially.
From genes to annotations